【恒兴大讲堂】新药研发过程中的药代动力学研究(三)
来源:恒兴医药

上篇我们聊到“如何从DMPK考虑新药成药性”,了解什么是成药性以及成药性评价中重要的两个经验法则,并对药物结构/理化特征与DMPK的关联性进行了一番探讨。那么在新药研发过程中,DMPK研究有些什么内容,而它又占据着什么地位呢?让我们继续往下看。
新药研发过程中的DMPK研究
临床前常规研究内容

可能的其他研究内容
1. 放射性同位素标记化合物的物质平衡、组织分布、血细胞结合、PK研究等;2. 微透析活体大脑取样研究(中枢神经系统药物);3. 离体皮肤渗透性研究(皮肤外用药);4. 游离药物PK研究(特殊血管内给药制剂);5. 眼部亚组织的药物渗透/PK研究(眼科药物);6. Unique metabolite(“独特”代谢产物)的动物PK;7. 主要代谢产物
临床前DMPK研究中,须充分利用体内体外的各种技术手段,力求完整地阐释药物本身的DMPK特性、并能从机理上较好地解释药物的毒性和药效特征(IND的药理毒理审评中通常注重结果的一致性及解读的合理性)。
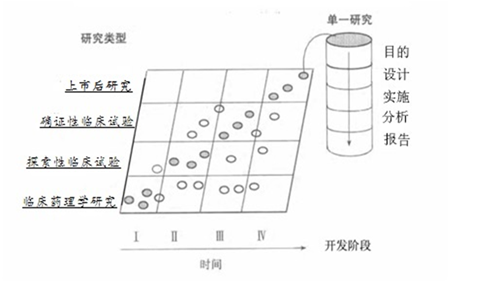
临床研究分类(按研究目的)
1)临床药理学研究:评价耐受性,明确并描述药代动力学及药效学特征,探索药物代谢和药物相互作用,以及评估药物活性。
2)探索性临床试验:探索目标适应症后续研究的给药方案,为有效性和安全性确证的研究设计、研究终点、方法学等提供基础。
3)确证性临床试验:确证有效性和安全性,为支持注册提供获益/风险关系评价基础,同时确定剂量与效应的关系。
4)上市后研究:改进对药物在普通人群、特殊人群和/或环境中的获益/风险关系的认识,发现少见不良反应,并为完善给药方案提供临床依据。
|
I期DMPK研究 (通常健康志愿者): |
II/III/IV期DMPK研究 |
|
耐受性研究(剂量爬坡)的伴随PK研究 |
特殊人群PK研究:肝功能损伤者、肾功能损伤者、老年人、儿童等 |
|
单次、多次给药PK研究 |
患者给药PK研究 |
|
食物对PK的研究(口服) |
不同人种PK研究 |
|
排泄和物质平衡 代谢产物的鉴定 药物相互作用(DDI) |
其它特殊目的的研究:剂量优化等 |
I期DMPK研究(通常健康志愿者):1)耐受性研究(剂量爬坡)的伴随PK研究;2)单次、多次给药PK研究;3)食物对PK的研究(口服);4)排泄和物质平衡;5)代谢产物的鉴定;6)药物相互作用(DDI)
Summary of Typical DMPK Assays/Experiments
In-vitro Studies(体外研究法)
①代谢稳定性(Metabolite ID and Profiling) ;②代谢酶表型(Reaction Phenotyping, CYP/UGT);③CYP酶抑制(CYP Inhibition);④CYP酶诱导(CYP Induction);⑤血浆蛋白结合(Plasma-Protein Binding);⑥全血/血浆比值(Blood/Plasma Conc. Ratio);⑦渗透性实验(Permeability Assay, Caco-2, PAMPA, MDCK)
In-vivo Studies(体内研究法)
①动物药动学(Animal PK);②组织分布(Tissue Distribution);③血脑屏障渗透(Blood-Brain Penetration, 脑组织/脑脊液/微透析);④排泄/物质平衡(Excreation/Mass Balance);⑤代谢物谱研究(Metabolite Profiling);⑥动物体内诱导(In Vivo Induction);⑦单向肠灌流(Single-Pass Intestinal Perfusion)
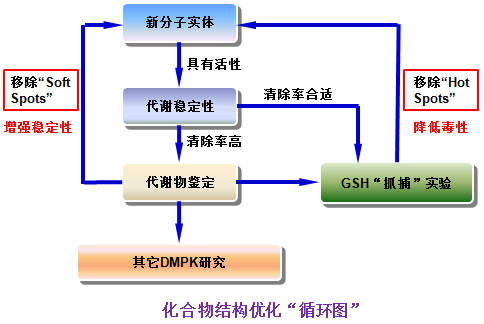
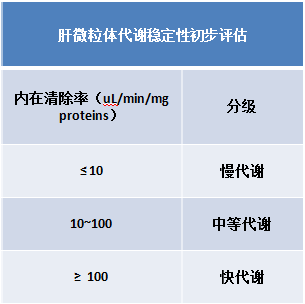
代谢稳定性及代谢物谱研究
代谢稳定性(Metabolic Stability)反映了化合物对生物转化的敏感性,它会影响化合物的口服生物利用度以及体内半衰期,进而影响到化合物在体内的安全性和有效性。
而利用体外代谢模型进行的代谢物谱研究(Metabolite Profiling),则可让科学家们可从代谢物的生成情况来进行化合物的优化、窥探药物分子可能具有的毒性和活性、并用以支持安全性评价的动物种属选择(主要焦点在犬和猴的选择上)。
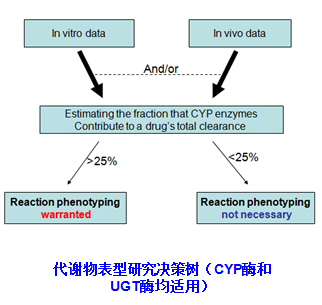
代谢酶表型研究
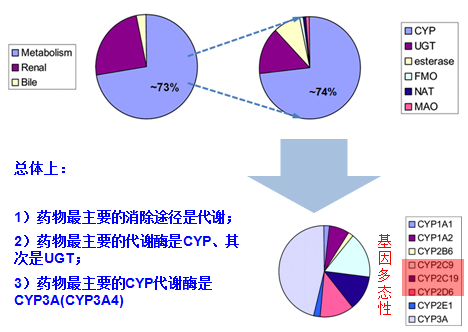
对于一个药物来说,了解参与其代谢的酶及这些酶的参与程度(即代谢表型研究)是非常重要的。这些信息可以帮助科学家们预见由于代谢酶的多态性而产生的个体间差异,并预测临床上药物间相互作用的可能性和程度。
CYP酶抑制与诱导
一个药物如果能抑制某代谢酶的活性(即抑制)或上调其表达(即诱导),其它为该代谢酶底物的同服药可能:1)代谢清除率降低、暴露量增加,从而造成安全性隐患;2)代谢清除率增加、暴露量降低,从而降低药效 。
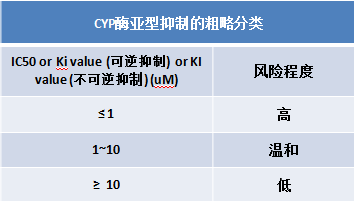
不可逆性抑制(大多情况下可等同于“时间依赖性抑制”和“机理性抑制”)造成的后果是最为严重的,药物如被发现为某主要代谢酶(如CYP3A)的强烈不可逆性抑制剂,会对其开发或临床应用造成“致命”的影响。

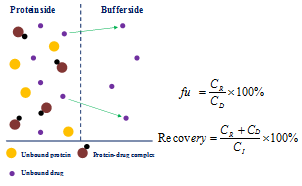
血浆蛋白结合与全血/血浆比值测定
一般认为,只有游离的药物才能在人体中发挥药效作用、被代谢转化和排泄。因此药物对血浆和组织蛋白的结合率会影响药物的药代动力学、药效和毒性。统计显示:已上市药物的血浆蛋白结合率25%、50%、75%分位数分别为约43%、83%及96%。
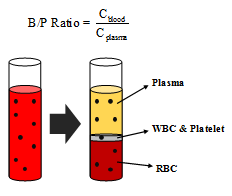
很多情况下我们需要的是“全血PK”,而不是“血浆PK”。因此,测定药物的全血/血浆浓度比对于正确理解药物的药代特性很有帮助。同时,全血-血浆浓度比可为潜在的血液毒性的评价提供参考。一般认为:大部分小分子药物的全血/血浆浓度比值在0.8~1.2之间。
CLblood = CLplasma/(Cblood/Cplasma)
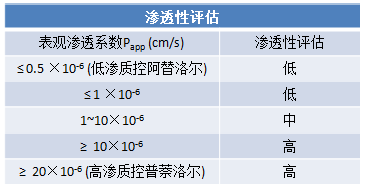
Caco-2 渗透性实验

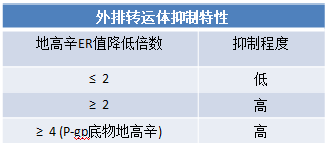
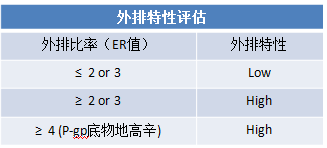
Caco-2细胞是一种人结肠癌细胞,在生长形成单层细胞膜后可较好地模拟小肠上皮细胞层(药物吸收部位)的特性。因此,而被广泛用作评估药物小肠渗透性的体外模型,同时Caco-2细胞也可表达不同的外排转运体如P-gp和BCRP等,因而也可用于药物外排转运体特性的研究。
科学家们希望看到的结果是:更高的表观渗透系数,外排比率接近于1.0、而不具有外排转运体抑制作用。
动物体内药动学研究
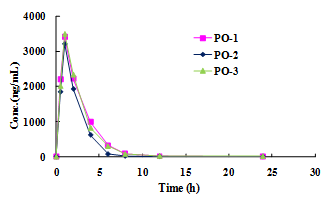
药物动力学(PK)是研究药物在体内随时间变化趋势和规律的科学。通常,科学家们先获得药物在容易获得的体液(如血浆或全血中)中的浓度经时曲线,然后利用数学模型对之进行研究以描述出药物在体内吸收、分布、代谢和排泄(ADME)的特征。一个典型的PK研究通常包括四个步骤:动物给药、样品收集、生物分析和PK参数计算。通常动物模型有:小鼠、大鼠、犬、猴。
良好的吸收及PK特性:1.良好的渗透性(Fa>85%)及生物利用度(如:F>60);2.最小的“首过效应”(肝提取率<30%);3.避免过早吸收(胃中吸收)或过快达峰(Tmax过小);4.药时曲线平缓,避免突升突降;5.较长的人体内半衰期(6-8h或更长);6.最小的食物影响或性别差异(临床给药方案的普适性);7.线性药动学(linear PK)
需尤为注意的是:在临床前研究阶段中,制剂通常不是最优的,这种情况下常常导致动物体内的生物利用度不理想。这种情况下,应进行合理评估,否则有可能“错杀”好的化合物。

一套设计良好的临床前DMPK申报内容应能:1)较全面地反映该药的药代特征,且DMPK研究数据之间具有一致性;2)较好、较全面地支持IND申报资料中的安全性和药效特征。
本期内容就到此结束了,不知道你们有什么看法呢?欢迎留言与我们讨论,那么下期“人体药动学预测的基本理论和实用方法”不见不散!
