【恒兴大讲堂】新药研发过程中的药代动力学研究(二)
来源:恒兴医药

好久不见!还记得上一篇的内容吗?不记得的赶紧去上一篇补补课吧,我们很快马上就要开始新的内容啦!
上一次我们讲到什么是药代动力学(DMPK),DMPK是一门研究药物分子在体内的存续和转化,及其与时间和空间关系的学科。换个角度说:是研究absorption(吸收)、distribution(分布)、metabolism(代谢)、excretion(排泄) 四个过程的。
“吸收”指的是从血管外途径给药后,药物分子进入到血液循环的过程。对“吸收”过程的描述,可分为“吸收速率”(理论上,以吸收速率常数Ka来表述)和“吸收程度” (理论上,以吸收比率Fa来表述)。注意:吸收不等于生物利用度(过于扩大化的理解),也不等于药物渗透(过于狭窄的理解)!其它的与吸收相关的参数/特性:生物利用度(F)、溶解性、渗透性。
药物最终的生物利用度,由药物的溶解、渗透、首过效应等诸多因素共同形成,可以如下公式表示:
F = (溶解比例) * (渗透比例) * (1-首过效应)
其中,吸收比率Fa = (溶解比例) * (渗透比例) ;而通常:(1-首过效应) ≈ (1-肝首过效应) = (1-Eh)。药物制剂主要以改变溶解性的方式对F产生影响,某些情况下(较少)也可以通过改变渗透性的方式产生影响。
注意:
1)药物分子要被溶解后才能被吸收;但溶解后的状态也极大地影响着吸收效率(分子状态比离子状态的跨细胞扩散效率高)。
2)生物利用度高,吸收比率一定高;生物利用度低,吸收比率不一定低(与制剂水平和给药剂量有很大关系)。
口服药物吸收的主要部位——小肠

小肠盘曲在腹腔内,全长4-6米,小肠粘膜形成许多环形皱褶和大量绒毛突入肠腔,每条绒毛的表面是一层柱状上皮细胞,柱状上皮细胞顶端的细胞膜又形成许多细小的突起,称微绒毛。小肠黏膜上的环形皱襞、小肠绒毛和每个小肠绒毛细胞游离面上的1000~3000根微绒毛,使小肠粘膜的表面积增加600倍,达到200平方米左右。另外,药物在小肠的驻留时间(residence time)长达3-5小时。尽管单位面积时间内的药物吸收量和吸收速率可能有限,但巨大的吸收面积和较长的驻留时间可极大地提高药物的吸收比率。
药物渗透的机制


药物分子在肠道渗透的四种方式(机制):跨细胞被动扩散(transcellular passive diffusion)、细胞旁被动扩散(paracellular passive diffusion)、主动载体运输 (active carrier-mediated transport)、促进载体扩散 (facilitated carrier-mediated diffusion)。前两种机制具有广泛性和较强的规律性,而后两种机制具有选择性。脂溶性强的分子(经验值:LogP>2),易通过跨细胞被动扩散来渗透;体积小的分子(经验值:MW<200),易通过细胞旁被动扩散来渗透。
药物分子被吸收进循环系统中后,将由动脉系统带至毛细血管,人身体有约100根毛细血管。在毛细血管处,血管内的物质与组织液中的物质(包括药物分子)进行充分的交换。
一般认为,MW< 1000 Da的物质可非常轻易地穿透血管壁,而分子量接近于或超过60KD的物质就非常难以透过血管壁了。几种可与药物分子结合的血浆蛋白分子量:蛋白67KD、α1-酸性糖蛋白40KD、脂蛋白则超过1000KD。
与药物分布相关的几个参数/特性:表观分布容积(Vd)、稳态表观分布容积(Vdss)、血浆蛋白结合率、组织蛋白结合率、Blood/Plasma比率、RBC/Plasma比率等。

人体(及动物体)针对外源性的异物(包括小分子和大分子药物)发展出了各种各样的“对抗”手段以将这些异物清除。清除的方式分为两种:一种是原形药直接“丢出国门”(排泄),另一种是将原形药“和平演变”(代谢)。
有公式:
CL (总) = CL (排泄) + CL (代谢)
Dose = A (排泄) + A (代谢)
A (排泄) /Dose (静脉) = CL (排泄) / CL (总)
A (代谢) /Dose (静脉) = CL (代谢) / CL (总)
人(及哺乳动物)最主要的代谢器官为肝脏,而最主要的代谢酶系为CYP(即P450)。
| 代谢器官 | 代谢酶系 |
|---|---|
|
肝脏(Liver)
小肠(Small intestine) 肺(Lung) 皮肤(Skin) 肾(Kidney) |
Cytochrome P450 (CYP) Flavin monooxygenase (FMO) Monoamine oxidase (MAO) UDP-glucronyltransferase (UGT) Sulfotransferase (SULT) N-acetyltransferase (NAT) |
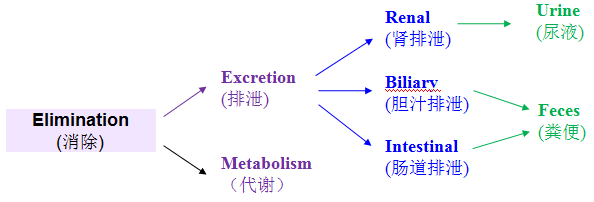
定义:药物从体内排出至体外的过程(排泄物可包括尿液、粪便、汗液、呼出的气体等)。
狭义的理解:原形药的排出过程;广义的理解:原形药和其代谢物的排出过程。大多数情况下特指原形药,但有时需具体情况具体理解。
排泄是药物(这里指原形药)消除的两种途径之一。而排泄途径主要包括三个:肾排泄、胆汁排泄、肠道排泄;最终主要排出在粪便和尿液中。绝大部分情况下,药物及代谢物主要都排出在尿液和粪便中,物质平衡的直接研究对象也主要是这两种。
如何从DMPK考虑新药成药性
成药性的基本意义:除了药理活性之外的可以让一个“化合物” 成为一个“药物候选物”或“真正的药”的其它特性。这些特性包罗广泛。
一般地,从科学的角度讲:成药性应包括适宜的理化、DMPK、安全性和药效等属性。尽管活性不属于成药性范畴,但“活性”要转化为“药效”却是需要DMPK/理化特性来支持的。举例:活性非常强,但半衰期太短,成药性差;活性非常强,但体内生成强毒性代谢产物,成药性差。
特别地,从经济性的角度讲:研究开发的便利性和低成本性。举例:脂溶性太强,制剂开发就难,成本增加;存在“独特”代谢物,就需专门针对该代谢物进行毒理研究,成本增加;对CYP3A4的抑制强烈,就需在临床开发中专门进行药物相互作用研究,成本增加;
从监管者的角度讲:科学问题解释的直接性、合理性和一致性。举例:设计中的前药如不能快速、彻底、专一地转化为目标活性物质,则对毒性的考虑会变得更复杂;DMPK数据无法解释毒理和药效;
其它方面:知识产权的合理性(举例:结构新颖性、是否突破现有专利)、临床用药便利性/顺应性(举例:半衰期太短,则服药频次高)。
-
良好的渗透性(Fa>85%)及生物利用度(如:F>60)
-
最小的“首过效应”(肝提取率<30%)
-
避免过早吸收(胃中吸收)或过快达峰(Tmax过小)
-
药时曲线平缓,避免突升突降
-
较长的人体内半衰期(6-8h或更长)
-
食物影响或性别差异小(临床给药方案的普适性)
-
线性药动学(linear PK)
-
个体间/个体内变异小
-
目标药效部位的适宜高度分布
-
非目标药效部位的最小分布
-
血细胞内外分布平衡(作用部位在血细胞除外)
-
血浆蛋白结合率不要太高(如高于99%将导致测定可靠性下降,进而极大影响到其使用价值;也使得蛋白结合引起的药物相互作用风险增大)
-
适宜的Vdss(Vdss<0.1L/kg意味着药物分子无法有效分布至血管外)
-
适宜的代谢稳定性(硬药/软药的概念)
-
无反应性代谢产物(reactive metabolites)
-
无药理活性代谢产物
-
多途径代谢(I相和II相、多种CYP亚型)
-
最小的多态性代谢酶(CYP2D6, 2C19)参与
-
最小的代谢酶抑制和诱导效应
-
动物与人代谢产物谱的一致性
-
代谢产物种类有限、集中度高
-
适宜的原形药排泄占比(胆汁、肠道排泄等),与另一消除途径代谢形成平衡
-
最小的药物外排特性(非外排药物转运体底物)
-
避免口服后短期内(如72小时内)大量的原形药在粪便中排出(意味着未吸收比例高和/或外排特性强)
-
冷物质回收率高(mass balance,指非同位素标记的原形药及其代谢产物)
总体上,达到“可接受”的DMPK特性即为成功
成药性评价上两个重要的经验法则:
一、Lipinski’s Rule of 5(五倍率法则)
-
分子量<500(体积特征)
-
氢键供体<5(作用力特征)
-
氢键受体<10(作用力特征)
-
LogP<5(脂溶性特征)
-
可旋转键≤10(刚性特征)
二、 Arup K. Ghose法则
-
-0.4<LogP <5.6(脂溶性特征)
-
40<Molar Refra.<130 (体积特征)
-
160<分子量<480(体积特征)
-
20<分子量<70(体积特征)
小分子药物的DMPK和理化特征本质上都是由其分子结构决定的。因此,DMPK和理化特征通常都与某些结构参数具有明显的相关性,而DMPK和理化特征之间也一定程度上存在着关联性。

LogP与血浆蛋白结合率之间具有显著的关联性
(作者本人未发表的研究成果,欢迎转图)
结构和理化参数与药物蛋白结合特点和体内分布特征的关联性。
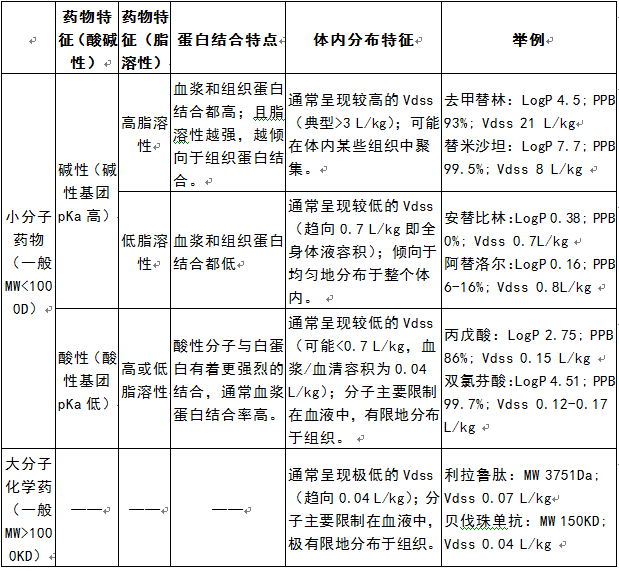
其他一些明显的关联性:分子量不高于800Da的小分子药物中,分子量越高、脂溶性越大的药物,通常越易被CYP酶所代谢、越易成为P-gp等外排转运体的底物、越难以通过肾脏排出至尿中。
从“什么是药代动力学(DMPK)”到“如何从DMPK考虑新药成药性”,作者分享了许多想法与理解,不知道你们有什么看法呢?欢迎留言讨论,那么我们下期“新药研发过程中的DMPK研究”不见不散!
